
产品中心
技术交流

扫描二维码
抗体开发20|抗体亲和力成熟结构改造的考虑
原创 Mabioway 文章来源:迈博睿
抗体通过其互补决定区 (CDR) 相互作用,从而赋予特定抗原的高度特异性。抗体的经典结构由四条链组成:两条相同的重链和两条相同的轻链,每条链在可变区的末端含有三个 CDR。其中,重链的第三个 CDR 通常最为复杂,在基因水平上通过 V(可变)、D(多样性)和 J(连接)基因的组合进行排列,这一过程称为 VDJ 重组。此外,这些区域特别容易发生体细胞超突变,这可以进一步扩展序列空间以引发针对病原体的免疫反应。当计算抗体序列空间的理论多样性时,组合似乎是无限的。用于治疗目的的抗体的常用发现工具往往依赖于各种技术,但大致可分为体内产生(例如动物免疫、杂交瘤或供体来源(例如 PBMC))或体外选择(例如噬菌体和酵母展示)。当面对外来病原体时,B 细胞表面会显示 1 型跨膜B 细胞受体,该受体通过 VDJ 重组作为亲和力成熟的前体。典型的估计是,人类平均携带 1012–1015个抗体序列。然而,体外选择的大小通常限制在 1010–1011,这仍然只是理论多样性的一小部分。这些技术的对比在于,免疫理论上可以利用无限的多样性,而体外文库需要额外的工程和多轮选择才能生成主要选定克隆的亲和力成熟变体。结构生物学的最新进展促进了抗体发现领域的发展,并为抗体-抗原相互作用提供了宝贵的见解。首先,蛋白质数据库目前已存储超过 160,000 个结构,其中近 4000 个结构属于抗体或抗体-抗原复合物。在原子水平上检查共结晶结构可以清楚地了解控制抗体-抗原相互作用的精确静电力和疏水力。我们将利用这些原理描述扫描抗体-抗原界面以调节这种相互作用亲和力的方法。在没有晶体结构的情况下,可以使用基于同源性和从头算结构预测方法洞察复杂结构,但主链和/或侧链构象预测的不确定性可能导致亲和力计算的不准确性。一般来说,模板和目标序列之间的序列同一性以及可用模板结构的质量是同源性建模中的重要考虑因素。同样,从头算结构预测的结果也受到方法、力场和接触势的选择的影响。将结合所提供的示例讨论将同源性建模纳入结构亲和力计算的敏感性。例如,我们将重点关注已发表的 DX-2930 亲和力成熟研究,DX-2930 是一种抗血浆激肽释放酶的抗体,该药物于 2018 年获得商业批准,用于预防性治疗遗传性血管性水肿 。有多种软件包可用于执行结构建模和亲和力计算,例如 MOE、BioLuminate 和 Rosetta。在本方法部分中,我们将重点介绍如何使用 MOE 。但是,许多一般原则都适用于这些不同的软件平台。1.基于结构的亲和力成熟:一个实例发现了一种抗血浆激肽释放酶 (pKal) 的抗体 M0162-A04,其亲和力为 5.39 nM。使用 VH-CDR3 内的摆动文库进行亲和力成熟活动,其中每个核苷酸有 85% 的机会保留为亲本,有 5% 的机会切换到其他三个核苷酸中的每一个。随后对变异文库进行噬菌体展示,产生了几个相对于亲本抗体具有改善亲和力的克隆(表1)。最终,克隆 M0199-A08 被选为最终候选药物。M0199-A08(也称为 DX-2930、lanadelumab)的晶体结构已在 PDB 中公布,既有 Fab(4PUB),也有与 pKal(4OGX、4OGY)的共晶体 。作为本分析的起点,基于 4OGX 模板构建了 M0162-A04 的同源性模型。如表1所示, M0162-A04 和 M0199-A08 之间的V H -CDR3中存在三处突变。此外,请注意,该示例旨在定义与抗原复合的整个抗体的残基相互作用,但讨论和分析将仅关注 V H -CDR3,因为已有关于该抗体的亲和力成熟数据的公布。应当注意,在本示例中,有些突变不会增加亲和力。相反,这些突变对亲和力要么有中性影响,要么有有害影响。
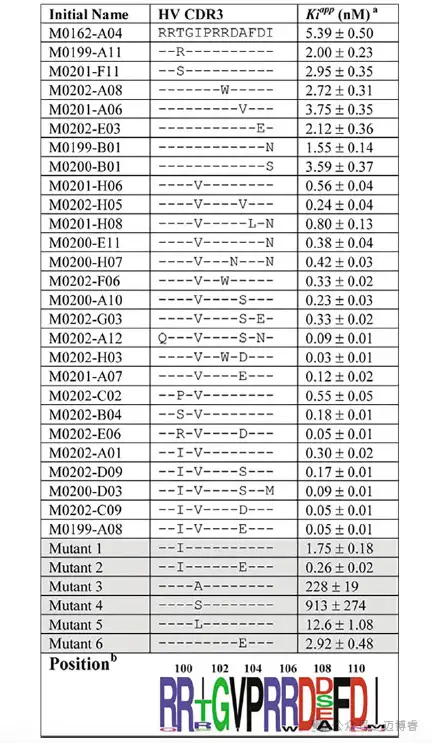
表 1 该表最初发表于《生物化学杂志》在 MOE 会话中,执行以下步骤:1.将共结晶抗体-抗原复合物装入 MOE。在此示例中,我们使用 M0162-A04 与 pKal 复合的同源模型。2.从下拉菜单栏中选择蛋白质 ➔ 结构准备。使用“纠正”按钮纠正三维坐标空间中任何缺失的原子。3.从 Structure Preparation 窗口中,选择 Protonate 3D 选项。通常,我们将 pH 值改为 6,盐浓度改为 0.0001,介电常数改为 4,溶剂介电常数为 80。我们通常使用 GB/VI 作为静电场,截止值为 15 埃,范德华模型为 800R3,非键合截止值为 10-12 埃。4.转到计算 ➔ 能量最小化。我们建议使用 Amber10:EHT 力场,将“开”设置为 10,“关”设置为 12。我们建议使用 Born 溶剂化模型,内部介电场为 4,外部介电场为 80。我们通常将结构最小化为最严格的梯度:0.00001 RMS kcal/mol/A 2。5.最小化完成后,通过选择注释➔抗体➔CCG来注释抗体和抗原。6.在蛋白质 ➔ 接触点下,选择抗体作为 A 组,抗原作为 B 组。搜索接触残基。完成后,单击顶行,滚动到底行,然后按住 Shift 键再次单击,突出显示窗口中的所有残基。从那里,选择“选择”按钮并打开 SEQ 窗口。取消选择抗原序列中的残基,然后选择 ➔ 设置创建,并将字段标记为“接口残基”。7.在蛋白质➔设计➔残基扫描下,默认选项为“活性链”,显示为“界面”。要将残基设置为仅抗体上有意义的残基,请在显示字段中选择“界面残基”,这将突出显示上一步中定义的抗体界面残基。8.默认突变列表将在突变字段中显示所有 20 种残基。可以将此列表编辑为较小的子集(例如,不包括 Cys),也可以将其扩展以包含非天然氨基酸。可以通过选择窗口中的所有残基,然后使用“应用”行右侧的记事本按钮选择/取消选择残基,轻松编辑整个残基块的此列表。或者,可以将输出文件从 prodesign.mdb 重命名为他们选择的名称。9.MOE 提供四种模拟运行选项。默认方法执行侧链包装和突变位点的能量最小化。另外三个选项采用集合平均值:低模式 MD、动力学和 Rotamer Explorer。此示例使用每种集合方法计算以进行比较。Rotamer Explorer 将执行侧链包装和能量最小化;但是,该方法使用侧链包装优化器中的解决方案样本,而不仅仅是最低能量包装。每个侧链配置都按照默认方法最小化。动力学选项运行分子动力学,突变位点侧链自由移动,突变骨架自由移动,附近残基骨架被束缚。低模式 MD 使用与动力学相同的束缚设置,但会过滤速度并专注于低模式运动(例如二面角扭曲),并避免高频振动。因此,它比 MD 更快地产生新构象。还建议选择亲和力选项来测量抗体和抗原之间的结合自由能的变化。10.这样就完成了残留扫描计算的设置。如果计算资源允许,请使用批处理功能创建可发送到高性能计算集群的 .svl 脚本。11.完成该作业后,将生成一个 MOE 数据库 (.mdb) 文件。在此文件中,将显示每个突变的亲和力和稳定性计算结果,以及每个突变体与亲本输入抗体相比的亲和力变化和稳定性变化、dAffinity 和 dStability。接受亲和力变化的阈值不是一成不变的,会根据预期构建的库大小和位置多样性而变化。在理想情况下,亲和力的提高将与负 dAffinity 相关。在此示例中,具有正确符号(负 dAffinity)变化的突变在表2中标记为绿色,在 0.5 kCal/mol 以内但具有错误符号(正 dAffinity)的突变突出显示为黄色,包含错误符号且大于 0.5 kCal/mol 的突变标记为红色。
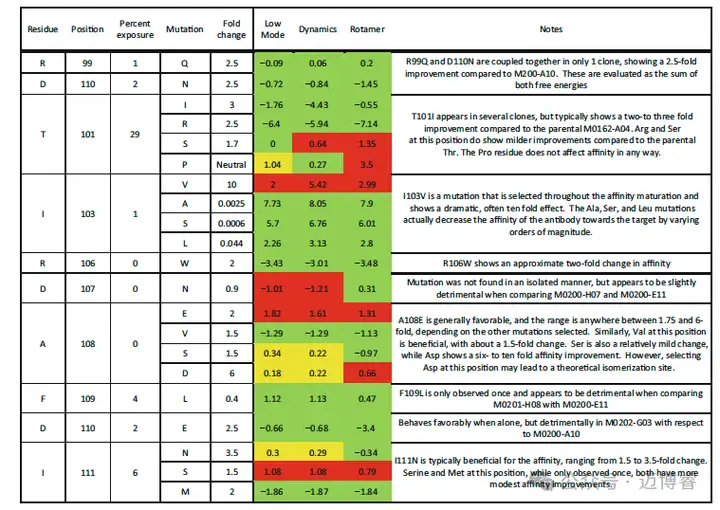
表 2 各突变对 pKal 亲和力总体变化倍数的影响总结2结果分析表2总结了Kenniston 等人表1中的亲和力数据,估计了每个氨基酸对亲和力倍数变化的贡献。一些突变具有很大程度的有效性,具体取决于序列中包含的其他突变。例如,表1中观察到两次 D110E 。在它的第一个实例中,在克隆 M0202-E03 中,它是唯一的突变,亲和力从 5.39 nM 增加到 2.12 nM,代表有利的相互作用,增加了 2.5 倍。D110E 的第二个实例发生在 M0202-G03 中,现在它与 I103V 和 A108S 突变相结合,该克隆的亲和力为 0.33 nM。然而,克隆 M0200-A10 仅包含 I103V 和 A108S 突变,其对 pKal 的亲和力为 0.23 nM。这意味着,当直接比较 M0200-A10 和 M0202-G03 时,D110E 对相互作用略有不利影响。此处尽可能在单个突变的背景下估计倍数变化。因此,突变 D110E 被评为有利。一对突变 R99Q 和 D110N 无法与其他突变分离。这些突变中的每一个仅被选择一次并出现在同一个克隆 (M0202-A12) 中。该克隆也可以直接与 M0200-A10 进行比较,并显示出更好的亲和力 (0.23–0.09 nM);因此,这两个点突变体的组合被认为是有利的,并且在本分析中,考虑它们的自由能总和,而不是它们各自的成分。表2中的数据显示,从定量角度来看,三种不同的残基扫描设计方法表现相当一致,这意味着自由能的符号在所有三种方法中都处于同一方向。将 KD 值转换为自由能变化后,我们发现计算的自由能变化量和实验的自由能变化量之间没有相关性。因此,我们只查看变化方向是否相同。大多数计算的自由能变化与测量的亲和力变化相关,但有几个明显的例外:I103V 和 A108E。在这里,我们将分析这些残基的预测,以及其他一些有趣的案例。如本实验描述所述,4OGX 的结晶复合物含有 I101、V103 和 E108。在进行残基扫描之前,这些残基分别通过同源建模恢复突变为 T101、I103 和 A108。这些同源建模的残基在三种残基扫描方法中显示出最大的自由能变化变化。例如,T101I 内的突变都显示出自由能的有利变化,与亲和力的增加很好地相关。然而,低模式、动力学和旋转体计算的值分别为 -1.76、-4.43 和 -0.55 kCal/mol,显示出很大的变化。I103V 突变体在三种方法中的计算结果分别为 +2、+5.42 和 +2.99 kCal/mol。不幸的是,这是子集中最重要的突变之一,因为仅此突变就导致亲和力增加了约 10 倍,从 5.39 nM 增加到 560 pM,因此此突变的 dAffinity 计算值非常正,这完全出乎意料。虽然 A108E 的变化幅度并不相同(亲和力仅增加了两倍),但计算结果还表明自由能变化的方向错误,三种计算方法的范围在 1.3 到 1.8 kCal/mol 之间。我们假设,与这些残基的同源性建模相关的细微变化是造成这些样品中自由能分布高度可变的原因,并且可以帮助解释实验观察到的亲和力变化与计算出的自由能值之间的差异。与更严格的自由能扰动方法相比,MMGBSA 方法的局限性也可能是另一个促成因素。第三,抗原中 CDR3 环和表位残基的移动性可能无法在这些计算中完全捕获。这些假设中的第一个很容易测试。我们准备了 4OGX 晶体结构,并使用低模式 MD 简单地执行从 DX-2930 到 M0162-A04 的突变,以计算 I101T、V103I 和 E108A 的自由能变化。这三种突变都应导致亲和力降低,因此应该对自由能产生正向变化。I101T (0.65 kCal/mol) 和 E108A (0.6 kCal/mol) 均同意这一假设,而 V103I (−0.3 kCal/mol) 实际上比使用同源模型残基时更接近于零。图1显示 I103 模型更适合 pKal 残基 478-480、Y555 和 W598 组成的口袋。考虑到异亮氨酸上的额外甲基有助于增加范德华接触,在这种情况下计算出的能量为 −0.3 kCal/mol 似乎是合理的。然而,亲和力的急剧变化很难从生物物理和计算的角度来理解。
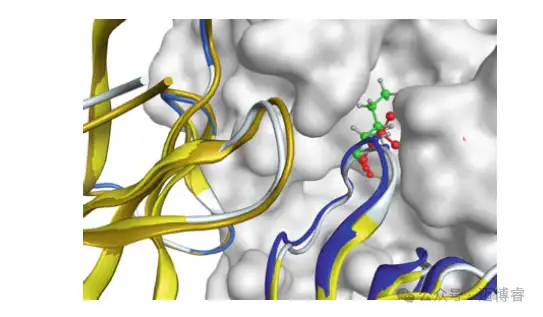
图1 同源模型中的 I103(绿色)与 4OGX 中的 V103(红色)的叠加类似地,T101I 比本研究中任何其他突变位点残基都更多地暴露于溶剂(29%),并且残基扫描方法对构象空间的采样程度可导致所报告的自由能变化范围很广。最后,A108E 位于 CDR3 的尾端,该位置被认为在结构上比 CDR3 内的大多数位置更稳定,这也许就是为什么这个位置的自由能变化没有太大变化的原因。在晶体结构中,E108 与 pKal 的 K575 相互作用。然而,在构建同源性模型时该相互作用被消除,并且在残基扫描期间从未重建;因此,使用该残基的计算是不正确的(图2)。
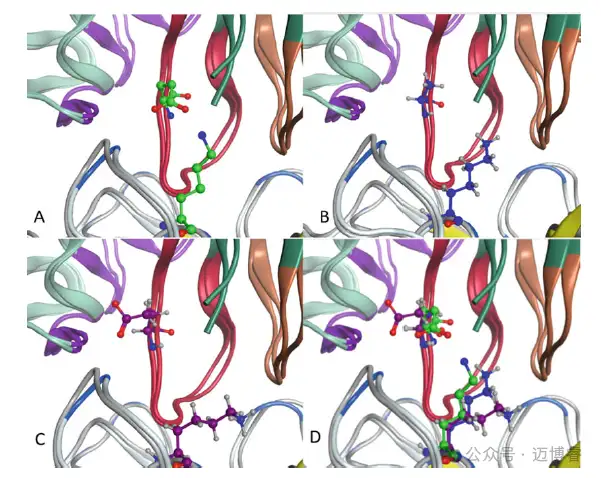
图2 DX-2930 E108 与未最小化共晶体结构中的血浆激肽释放酶 K575 相互作用 ( a )。同源性建模和最小化后的抗体 A108 ( b )。转换为 E108 后低模式 MD 分析的输出构象 ( c )。a 、b和c的叠加显示,E108 和 K575 的天然相互作用被破坏,因为在这些模拟中 K575 的轨迹向 pKal 结构回缩,从建模和最小化开始短暂,但在残留扫描期间加速尽管从亲和力未成熟亲本的同源模型重建 DX-2930 存在这些显著的失败,但这些数据中还是有几个成功案例。在位置 103,Ala、Ser 和 Leu 是经过改造的变体,旨在降低抗体对 pKal 的亲和力,并且这些残基中的每一个都显示出强烈的正向自由能变化。然而,使用同源模型不利于自由能计算的逻辑,还使用 4OGX 晶体结构中 V103 的低模式 MD 计算了这三个突变的 dAffinity。在这里,与残基相关的自由能变化为 Ala +4.78 kCal/mol、Leu +1.12 kCal/mol 和 Ser +0.16 kCal/mol。此位置的丙氨酸将亲和力降低至 228 nM,这与自由能的大幅变化很好地相关。同样,L103 对应的亲和力略微下降至 12 nM,反映出突变后自由能略微增加。另一方面,丝氨酸的亲和力变化最为剧烈(913 nM),但自由能计算并未反映出这一点,因为这种突变被认为在能量上与 V103 一致。3使用计算机计算生成库如本例所示,亲和力成熟的输入是描述两种蛋白质之间相互作用的结构,然后进行计算机残基扫描,从而输出突变及其相关的结合自由能变化。对于上一节中描述的工作示例,小于零的自由能变化通常预示着有助于增加亲和力的替换,而大于零的自由能变化通常表示突变降低了两种蛋白质的亲和力。下一个挑战是评估计算机突变分析的输出并生成一个要淘选的物理文库,这需要了解文库大小和淘选方法的限制。在上面描述的例子中,66 个 CDR 残基中有 46 个与 pKal 接触。按 dAffinity 排序,并取 -0.5 kCal/mol 的任意截止值,共有 233 个推定的突变,平均每个位点约有 5 个突变,不包括亲本残基(数据未显示)。基于这些近似值,理论文库多样性估计为 6 46,相当于 6.2 × 10 35的大小。鉴于典型噬菌体文库的大小在 10 10的数量级,这个数字是天文数字,无法物理合成用于淘选,而且这个文库的采样将非常差。然而,在设计文库时,其他考虑因素(例如预先选择抗体-抗原界面或 CDR 上的较少位置、最大限度地利用种系残基、可开发性以及消除潜在的化学降解倾向)可以大大减少计算设计文库的理论大小(稍后介绍)。例如,使用一般对抗网络设计的文库能够同时考虑可开发性和抗原结合。此外,在计划构建亲和力成熟文库时,还应考虑其他实验策略。合成各种 DNA 寡核苷酸池已成为抗体亲和力成熟的常用技术,并且在过去十年中,这些池的创建精度已大大提高。这些服务在大多数 DNA 合成供应商中很常见,不同的技术对单个 DNA 链合成精度的控制程度不同。对这些单个技术的评估超出了本章的范围。为了更深入地探究理论上的多样性,可以针对每个单独的 CDR 构建文库以筛选亲和力更高的结合物。此过程专门用于生成 DX-2930,如上例所示,其中文库专门针对 VH - CDR3(尽管此例中的文库是使用核苷酸序列的加权随机化生成的,而不是受到计算机预测的影响)。尽管如此,为每个 CDR 和 CDR 组合(例如整条重链或整条轻链)创建文库是筛选亲和力增加的常用技术。这些单独选择的输出可以与标准分子生物学过程相结合,并且可以生成一个新的文库,其潜力甚至比单个组件文库实现更高的亲和力。从另一个角度来看,可以通过限制某些残基组合来降低文库的复杂性,特别是那些可能导致假定缺陷的组合,例如氧化、脱酰胺、异构化和糖基化。通过避免这些基序来减少理论多样性有时可以大大减少搜索空间。本书、Thorsteinson 撰写的一章以及文献全面回顾了基于计算机的可开发性方法。最后,还可以通过对计算出的自由能变化设置更严格的过滤器来降低库的复杂性。如果一个(或多个)CDR 具有大量与抗原相互作用的残基,这可能尤为重要。相反,可以对仅与抗原形成几个相互作用的 CDR 应用较不严格的过滤器,从而在该区域内产生更大的多样性。文库构建是一门艺术和一门科学之间的平衡,没有一种亲和力成熟策略能够广泛涵盖与改善抗体与其抗原之间的相互作用相关的理论多样性。然而,我们希望本节能为采样广阔的序列空间提供一些合理的见解,以期提高抗体与其抗原之间的亲和力。4未来方向亲和力成熟是优化新发现的先导候选物过程中的一个重要步骤。该分析表明,科学家可以合理地定义突变集,以创建一个变体库,这将有助于提高抗体-抗原相互作用的亲和力。执行此类任务的关键要求之一是抗体和抗原之间的共结晶复合物。正如本例中观察到的,该方法的可靠性需要精确布局两种蛋白质之间的原子相互作用。可以使用同源性建模,但如果 CDR 中各个氨基酸的主链和侧链旋转异构体构象没有精确定义,结果可能会变得不那么可靠。此外,目视检查原子接触可以帮助引导残基并入库中。对于本章中的示例,在重新创建从 M0162-A04 亲和力成熟库中选择的点突变时存在挑战。当然,与在开始此练习之前模拟三个回复突变相比,拥有 M0162-A04 和血浆激肽释放酶的共结晶复合物可能有助于阐明这两种蛋白质之间的接触。特别是,导致亲和力增加十倍的 I103V 突变可能仍然难以预测。然而,该方法表明,生成可以提高亲和力的库实际上是相当可行的,尤其是当引入其他在计算机中生成的能量有利突变时,但由于未在表1中列出,因此不属于此分析的一部分。由于该方法需要精确的分子相互作用来进行自由能计算,因此这一新兴领域的未来方向很明确:抗体-抗原复合物预测的改进可以大大提高计算机计算的有效性,从而实现更好的库设计。随着结构预测的改进,本章中的方法可以应用于由抗体和抗原的蛋白质-蛋白质对接同源性模型产生的预测抗体-抗原复合物,以生成计算机亲和力成熟库。
【关于逐典】
上海逐典生物科技有限公司,坐落于中国(上海)自由贸易试验区,获得ISO9001质量体系认证,是一家从事重组蛋白研发和销售的高新科技企业。
逐典生物始终秉持以客户为中心的理念,针对重组蛋白的结构设计、纯化工艺及其稳定剂型相关的多项关键技术进行优化。专业定向蛋白变复性技术,可将大肠杆菌大量表达的变性固体蛋白转变成高活性可溶性蛋白。凭借技术优势,逐典生物新品研发周期短且可控性强,为重组蛋白的高质高效研发提供保障,为企业生产降本增效。
公司自成立以来成功开发百余种高活性细胞因子及多种高活性蛋白酶,覆盖细胞培养、病毒纯化以及质量分析等生物工艺各个环节。可广泛应用于科研、医药生产及IVD(体外诊断试剂)等领域,满足各类用户所需。

 粤公网安备 44010602000473号
粤ICP备09063491号
营业执照
粤公网安备 44010602000473号
粤ICP备09063491号
营业执照

