
产品中心
技术交流

扫描二维码
当我们讨论LVV in vivo delivery platform 我们到底在讨论什么 Carl June 综述衍生
文章来源公众号:VTALK 作者:VTALK

第 1 部分:技术格局分析
1.1平台对比矩阵
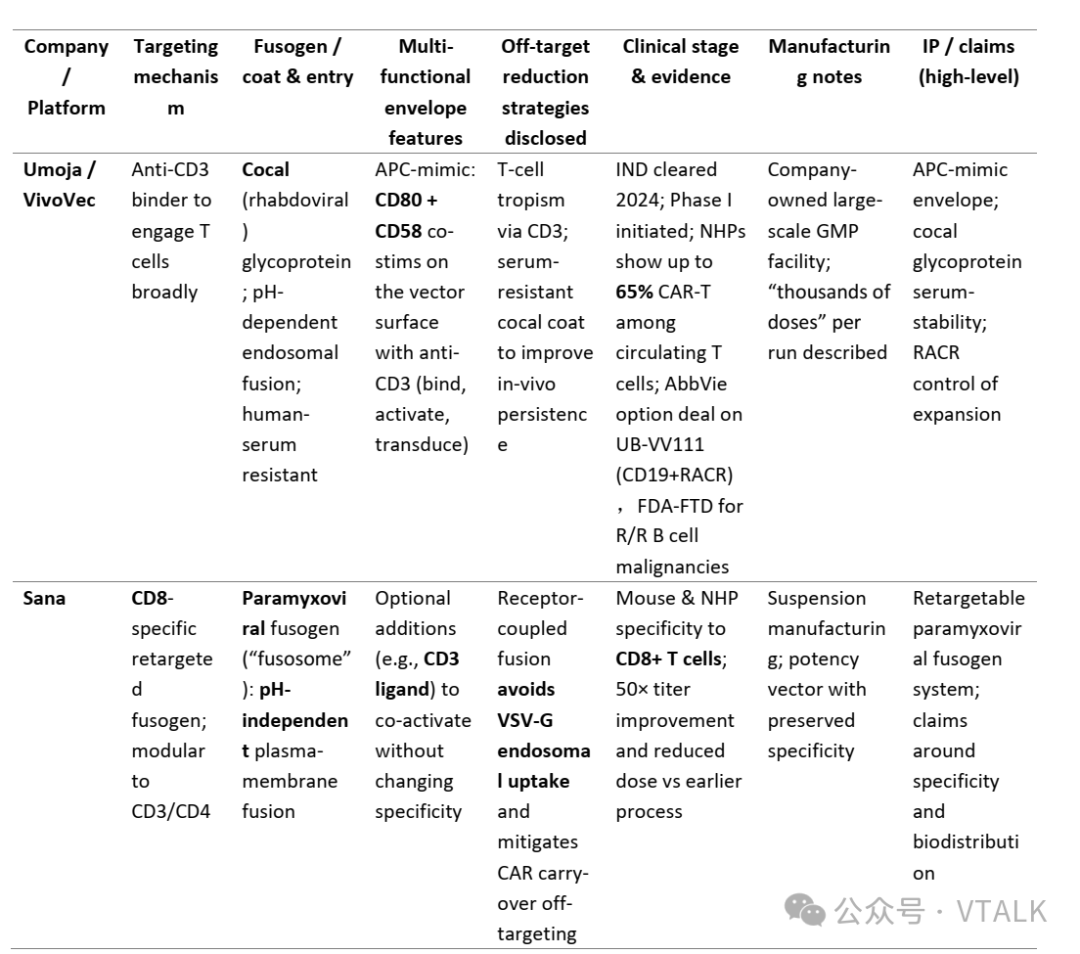
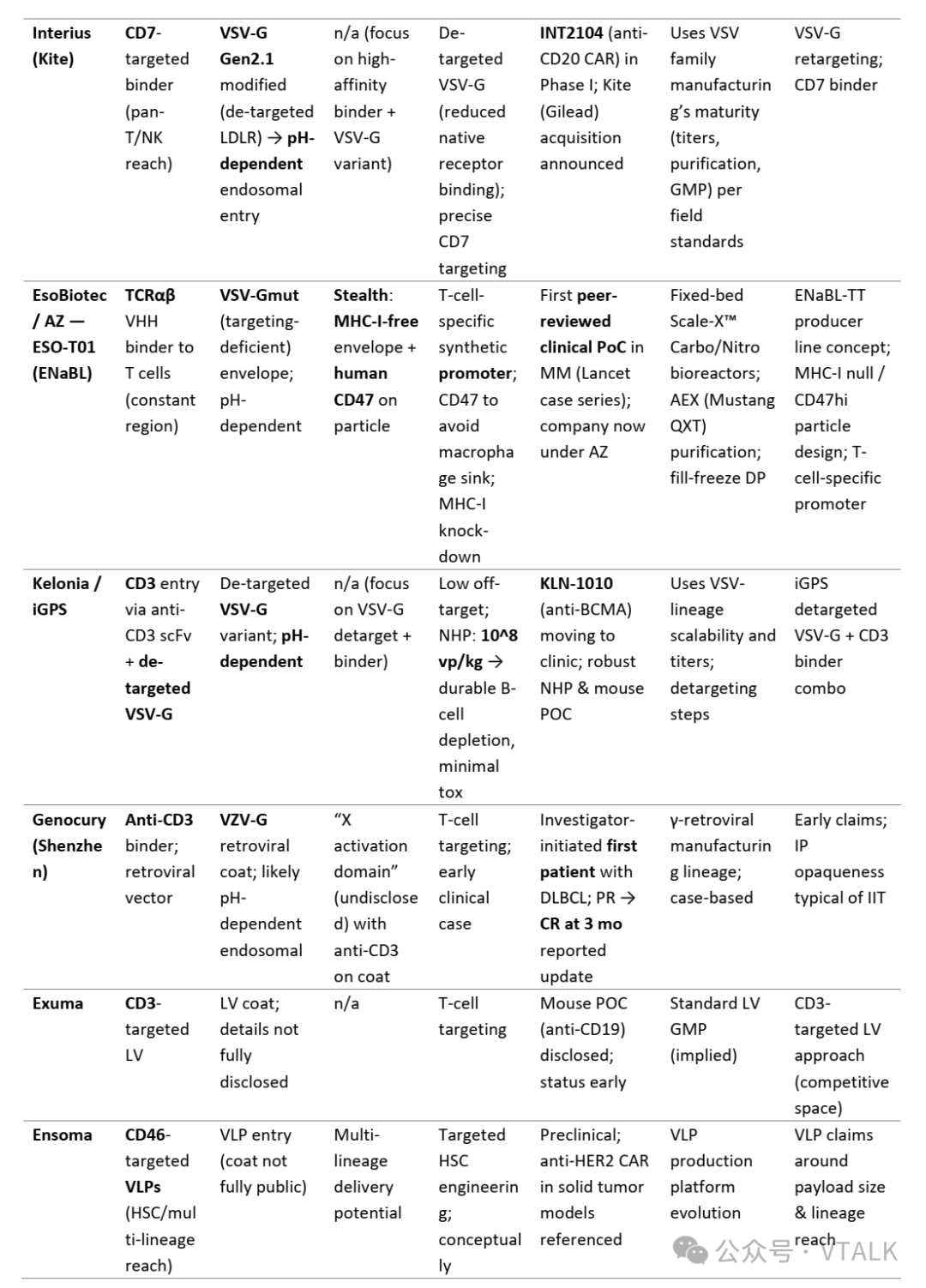
注:update to 2025。
Kelonia Therapeutics 正在开发一种创新的体内基因编辑系统 (iGPS) 平台,用于直接在患者体内生成 CAR-T 细胞。 iGPS 平台是通过复杂的病毒载体工程开发的:
·去靶向天然受体:对 VSV-G(水疱性口炎病毒糖蛋白)包膜进行修饰,以消除与其天然受体 LDL-R(低密度脂蛋白受体)的结合
·T 细胞特异性靶向:掺入抗 CD3 单链可变片段 (scFv) 以为 T 细胞提供特异性趋向性
·优化过程:筛选了多个 VSV-G 变体,以确定那些:
·最佳抑制 LDL-R 结合
·保持有效的 T 细胞转导能力

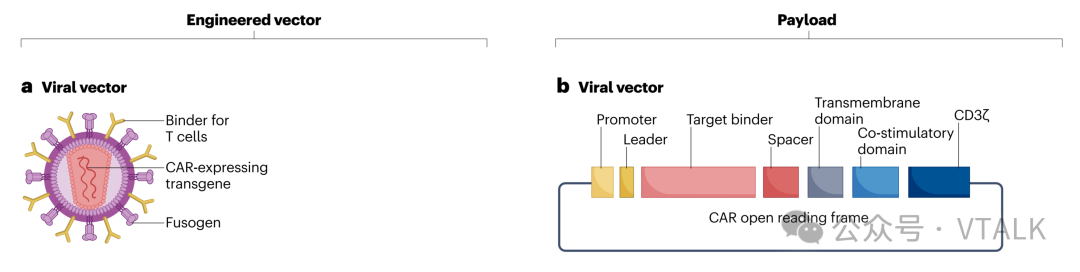
1.2 技术准备评估
·临床验证:ESO-T01 (BCMA) — 《柳叶刀》上第一个经过同行评审的体内 CAR-T 病例系列 (RRMM);ENaBL颗粒设计(MHC-I–/CD47+/TCRαβ VHH;T细胞特异性启动子)在临床补充剂中公开。
·临床(I 期活性/启动)阶段:Umoja (CD19+RACR) 和 Interius (INT2104,CD7 靶向抗 CD20)——均已进入人体临床研究。Kite 宣布有意收购 Interius; 艾伯维选择了 Umoja 的 UB-VV111进行开发合作,UB-VV111于日前获得FDA的FTD(fast track designation)。
·临床前阶段:Sana — 具有 NHP 特异性的 CD8 靶向副粘病毒融合体系统(Paramyxoviral),与早期构建的fusogen相比,滴度更高,能够减少剂量; Kelonia — 初步展现的 NHP 药理数据和计划于 2025 年公布的去靶向 VSV-G CD3 管线。
·早期临床前:Exuma、Ensoma (VLP 多谱系体内工程)和其他进入者。
·
第二部分:策略优劣势分析
2.1 靶向策略分析(CD3、CD4、CD8、CD7、TCRαβ)
·CD3
o优点: 广泛的泛-T摄取(CD4/CD8),强大的激活协同作用(例如,Umoja的CD3 + CD80 / CD58 APC模拟物);在临床前/大型动物环境中的稳健转导。
o缺点: 如果激活太强,更广泛的趋向性会增加细胞因子激增的风险;如果没有护栏,则有可能转导 T 细胞肿瘤中的恶性 T 细胞。
o治疗指标: 有利于快速 T 细胞扩增;使用 VivoVec 观察到循环 T 细胞的 NHP 峰值高达 ~65% CAR-T。
o定位: 较多玩家的竞争(Umoja、Kelonia、Genocury、Exuma)——通过APC 模拟与隐身功能和安全导轨进行区分。
·CD4
o优点: 辅助性 T 细胞偏倚;在某些情况下可以避免恶性 CD8+ 克隆的转导。
o缺点:CD4+ T 细胞恶性肿瘤的风险;辅助偏倚可能会降低直接细胞毒性分数。
o证据: 副粘病毒 NiV 重定向框架允许在临床前工作中进行 CD4 特异性转导。
·CD8
o优点:选择性递送至细胞毒性T细胞;Sana CD8 fusogen的临床前/NHP数据显示出很强的特异性和淋巴器官靶向性;可避免在T细胞恶性肿瘤中将CAR递送至CD4+恶性细胞。
o缺点:无辅助T细胞支持,可能需要共同刺激或细胞因子支持才能持续存在。
o定位: 对B细胞癌具有很强的拟合性,具有安全优势;利用副粘病毒 pH值不依赖的 进入优势(第2.2节)。
·CD7
o优点:广泛的 T/NK 覆盖范围 (Interius),支持多效应器 CAR 种群。去靶向的 VSV-G 变体 (Gen2.1) 增强持久性并减少意外的 LDLR 结合。
o缺点: 正常 T/NK 中 CD7 抗原表达增加了复杂性;离体潜在的自相残杀(When a CAR targets a T-cell antigen (e.g., CD7, CD5, TRBC1/2), engineered T cells kill themselves or each other during manufacturing because they express the same antigen they’re meant to attack. Result: poor cell yields, unstable products, or the need for edits/PEBLs/knockdowns to remove)不适用于此,但生物学仍然相关;需要仔细的安全门控。
·TCRαβ(恒定区)
o优点: 靶向常规 αβ T 细胞,无论子集如何;与 MHC-I–/CD47+ 隐形涂层和 T 细胞特异性 启动子 (ESO-T01) 配对以限制非 T 表达。
o缺点: Pan-T 与 CD3 一样到达;在很大程度上依赖 启动子特异性和 包膜隐蔽性来防止脱靶转导和先天清除。
o临床信号:通过该设计实现的第一个经过同行评审的人体疗效/安全性信号 (Esobiotech)(RRMM)。


2.2 Fusogen进入机制(pH 依赖性 VSV 谱系与 pH 依赖性副粘病毒)
·特异性和脱靶
o副粘病毒(NiV/麻疹衍生):质膜融合与受体结合偶联,→体内 VSV-G 家族可见的更高的靶向特异性和减少的非特异性内体摄取。
oVSV 谱系(VSV-G / cocal 变体): 高滴度和成熟的 GMP,但 pH 依赖性 内体融合可以允许 非特异性摄取; 缓解措施包括 去靶向 VSV-G(通过引入多个具有氨基酸取代的 VSV-G 变体,例如 Interius Gen2.1)和血清耐药 cocal(Umoja Biopharma 的 VivoVec 平台 使用 Cocal 病毒融合糖蛋白 而不是传统的包膜蛋白)。
·CMC挑战
oVSV 谱系:通常滴度最高,规模稳健,纯化成熟;有吸引力的 COG。
o副粘病毒:历史上较低的滴度;Sana 报告称,滴度/工艺进步 50×,可在保持特异性的同时减少剂量。
·临床应用特征
oVSV 系列现在利用对 GMP 的熟悉度,有多个已经进入临床开发阶段的LVV in vivo CAR T产品(Umoja、Interius)。
o副粘病毒 CD8 fusogen 平台具有 NHP 特异性和生物分布优势(报告肝脏信号降低),支持进一步开发。
·创新潜力
o副粘病毒系统对于受体重定向具有高度模块化;VSV 谱系可以去靶向工程化处理(Kelonia、Interius)结合。
2.3 多功能Envelop工程
·APC 模拟 (Umoja):载体显示抗 CD3 + CD80/CD58,在一个颗粒中提供结合 + 激活 + 转导 — 加速 T 细胞原位启动,但提出了细胞因子管理考虑因素。
·免疫隐身(EsoBiotec):MHC-I-包膜加CD47逃避吞噬作用,降低清除率/先天摄取;与T细胞特异性启动子配对抑制非T表达。
·多谱系(Ensoma VLP): 靶向 CD46 的 VLP 旨在 HSC 产生多谱系效应子;CAR-T 处于早期阶段,但有可能扩展到 T 细胞之外。
权衡:增加的envelope功能增加了 CMC 的复杂性(分析、一致性),而不是明显的治疗益处(快速激活与隐身和持久性)。Umoja 和 EsoBiotec 说明了不同的、可防御的护城河(APC 模拟与隐形启动子+CD47)。
2.4 脱靶效应减少策略
ü受体靶向fusogen/binding domain(CD8/CD3/CD7/TCRαβ) — 特异性的主要驱动因素。
ü副粘病毒进入(受体偶联,非pH依赖),以避免 VSV-G 出现的内体非特异性摄取。
ü去靶向的 VSV-G + 结合剂 (Kelonia、Interius)可减少天然 LDLR 结合;保留强滴度。
ü包膜上的 CD47 (和 MHC-I 减少)以减少 吞噬作用汇 和先天摄取(ESO-T01;也普遍用于 LV)。
üT 细胞特异性启动子(以及 2025 年摘要报告中的新型合成启动子)以最大限度地减少 整合后的非 T 表达。
ü防止载体颗粒上的 CAR 残留 (VSV-G 的主要脱靶风险):2025 年“第四代”LVV基因组 + TRiP + 2KO 消除了颗粒中可检测的 CAR ,并减少了异常的 vRNA 剪接。
üCAR 残留是指在制造过程中慢病毒 (LV) 颗粒上/中意外存在 CAR 蛋白(和/或编码 CAR 的剪接病毒 RNA)。当这些颗粒用 VSV-G 进行假分型时,病毒粒子表面的 CAR 可以将其抗原结合在非 T 细胞上并重新靶向载体,从而驱动脱靶转导,而不受预期的融合原趋向性的影响。这种现象已被明确标记为 VSV-G 系统的安全风险:CAR 蛋白可以在 LV 生产过程中掺入载体颗粒中,并且当与 VSV-G 融合剂结合时,已被证明可以驱动脱靶转导。
ü重要性:向携带抗原的旁观者细胞(例如 B 细胞、髓系细胞或肿瘤组织)脱靶递送会产生可避免的插入风险,并可能混淆生物分布、效力和安全性读数。pH 依赖性内体进入途径(VSV-G 谱系)已经容易发生非特异性摄取;在颗粒上添加 CAR 会进一步破坏特异性。
ü2025年“第四代”修复:2KO+TRiP(带TRAP/tbs):
ü2KO 基因组:第四代LVV基因组 (SupA2KO-LV),可灭活 MSD 驱动的剪接,消除那些编码 CAR 的剪接 vRNA。
üTRiP系统:将TRAP结合序列(tbs)插入CAR 5′ UTR(与Kozak / ATG重叠)并在 生产者系中表达TRAP;TRAP 结合结核病并抑制生产细胞中的 CAR 翻译,防止 CAR 蛋白出现在生产者膜上(从而出现在出芽颗粒上)。
ü使用该第四代构建物重靶 NiV-糖蛋白 LV(pH 不依赖入口)显示出更高的 T 细胞特异性, 并 避免了 VSV-G 载体在体内看到的脱靶信号(例如,骨髓/肝脏摄取)。
·药物联合治疗:目标:在LVV给药时瞬时提高靶向转导和安全性,而不影响以后的CAR-T扩增/功能。
ü两个互补boost:
l雷帕霉素(mTORC1 抑制剂)→改善 T 细胞内的载体进入/运输。
l达沙替尼 (Src/Lck 酪氨酸激酶抑制剂)→ 抑制 TCR 信号传导,阻止 CD3 内化,减少急性细胞因子释放,并可以暂时“暂停”CAR 活性。
ü
第 3 部分:竞争格局和市场定位
3.1 临床证据层次结构
一.ESO-T01 / AZ — 第一个经过同行评审的 MM 临床 PoC;设计包括 MHC-I–/CD47+ 包膜和 T 细胞特异性启动子。
二.遗传学 — 使用VZV-G逆转录病毒载体和抗CD3结合剂的IIT首次患者报告(iit first-patients report, DLBCL );PR → CR 更新 3 个月。
三.Umoja — I 期活性 (CD19+RACR);强大的 NHP 经验;艾伯维选项验证。
四.Interius (Kite) — CD7 靶向 LV + 改良 VSV-G 的 I 期;Kite 的收购表明对平台的信念。
五.Kelonia — 晚期临床前,NHP B 细胞耗竭强劲,浓度为 10^8 vp/kg,毒性低; IND 就。
六.Sana — 具有 NHP CD8 靶向和特异性生物分布的高级临床前; 制造效率提高。
七.Ensoma/Exuma — 早期临床前(分别为VLP和CD3-LV)。
3.2 适应症策略拟合度
·血液系统恶性肿瘤(B 细胞):跨平台最强拟合; CD19/22,由 NHP 和早期临床信号(Umoja、Interius、Kelonia、ESO-T01)支持的 BCMA 靶点。
·T细胞恶性肿瘤:CD8靶向 方法有助于避免转导恶性CD4细胞;存在临床前支持。
·实体瘤: 早期(Ensoma 抗 HER2 VLP 临床前);TME 障碍仍然存在;可能需要装甲/连击。
·自身免疫: 兴趣日益浓厚(例如,自身免疫的Interius路线图);安全标准高;启动子/包膜特异性至关重要。
3.3 未满足的需求和空白
·VSV 谱系系统的稳健脱靶硬化(脱靶向之外)→采用 TRiP/2KO 基因组和启动子对照。
·肝脏和巨噬细胞 → CD47 和抗吞噬涂层;进一步的生物分布调整。
·用于适应症定制的子集选择性递送(CD8 与 CD3/CD7); 双靶点有效载荷以减轻抗原逃逸。
·实体瘤铠甲装配和 组合 策略(检查点/趋化因子)仍处于早期体内形式。

第 4 部分:关键挑战和风险评估
4.1 安全
·CRS/ICANS: 激活前向包膜 (APC-mimic) 需要仔细剂量和托珠单抗/标准措施(见于 NHP)。
·插入诱变: 根据综合 LV 标准(监管预期)进行长期随访。
·长期的靶向效应: CD19/BCMA 计划预计会出现 B 细胞发育不全(根据需要使用 Ig 替代疗法进行管理)。
·免疫原性和再给药: 包膜/结合剂免疫原性和抗载体免疫需求计划(副粘病毒与 VSV 谱系贸易)。
4.2 功效
·转导变异性:副粘病毒受体偶联进入提高了 特异性; 混合包膜 NiV(75% 重靶向 + 25% 非靶向)可提高 T 细胞进入和效率。
·扩展和持久性: VivoVec 在 NHP 中显示出强劲的峰值和二次扩展;RACR 提供药物控制。
·抗原逃逸: 考虑LVV cargo中的早期双抗原(例如 CD19+CD22)设计。
·TME(实体瘤): 挑战较大
4.3 制造与可扩展性
·滴度/产量: VSV 谱系最高;副粘病毒滴度 改善 (Sana)。
·血清稳定性和配方:Cocal 涂层支持直接输液; 隐身 功能。
·GMP 复杂性: 多域包络和工程生产线 (ENaBL-TT) 增加了分析/控制需求。
·规模:Umoja已经进行规模化生产的准备,从自体细胞治疗到真正现货型治疗是质的飞跃
 粤公网安备 44010602000473号
粤ICP备09063491号
营业执照
粤公网安备 44010602000473号
粤ICP备09063491号
营业执照

