
产品中心
技术交流

扫描二维码
处于临床研究阶段的PROTACs
本文来源于:新药新发现
蛋白水解靶向嵌合体 (PROTAC) 是一种新兴的靶向降解蛋白质的治疗策略,它通过将目标蛋白(POI)带至 E3 连接酶附近,利用泛素蛋白酶体系统 (UPS) 来促进蛋白质的降解。相比传统的抑制蛋白活性,PROTAC 能够选择性地从细胞中清除特定蛋白,提供了事件驱动的降解机制,避免了单纯的占用率依赖。药理学上,PROTAC 有助于更彻底地阻断蛋白功能,延长作用时间,并可能模拟基因敲除的效果。由于三元复合物的形成依赖于 POI 和 E3 的特异性相互作用,PROTAC 能够选择性地作用于特定异构体和组织。研究表明,PROTAC 已在临床中显示出良好的耐受性和疗效,目前正在后期临床试验中。阿斯利康概述了 PROTAC 各组成部分的优化以及药物化学家在这一充满挑战的领域寻求实现口服生物利用度所面临的困难。并基于性质,总结 PROTAC 优化的方法。
01
Clinical PROTACs
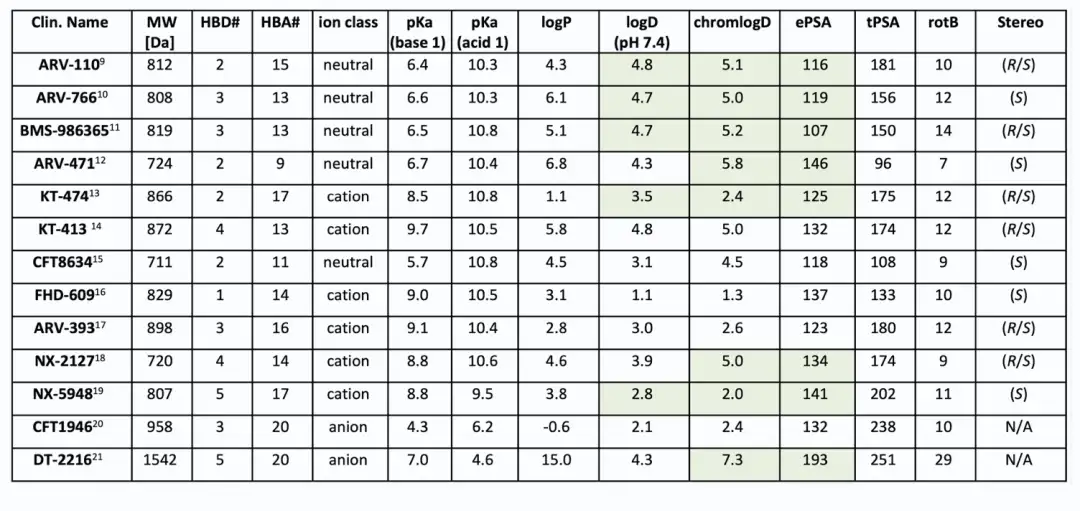
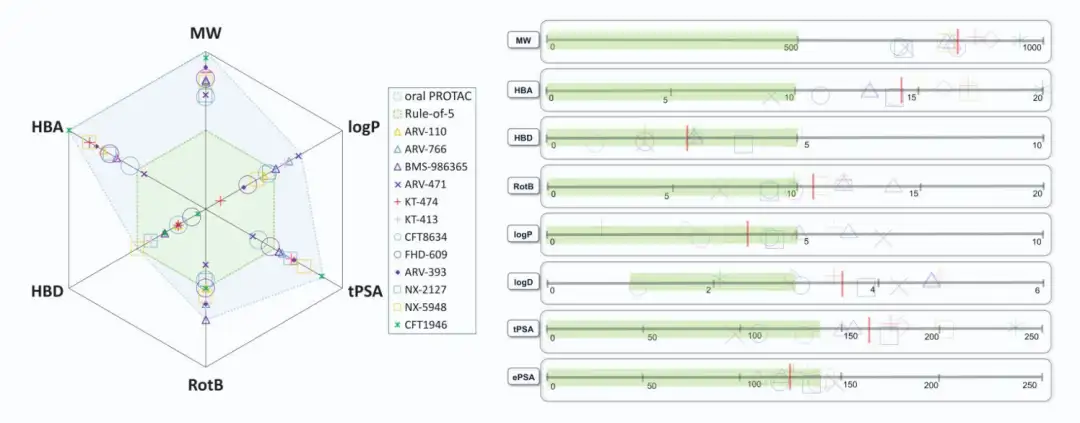
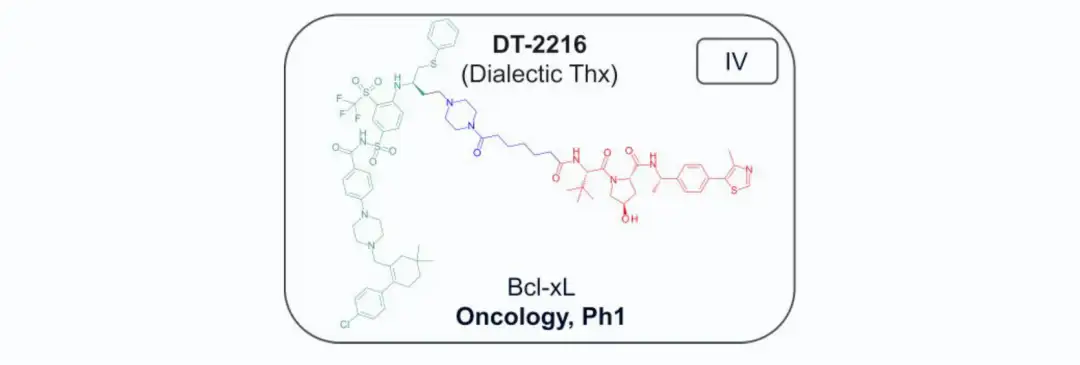
截至2024年7月,共有13种结构公开的蛋白水解靶向嵌合体(PROTAC)进入临床试验,主要用于肿瘤治疗,其中一种用于炎症治疗。两种PROTAC(ARV-110和KT-413)因战略原因停产。在这些化合物中,12种与E3连接酶cereblon(CRBN)结合,只有DT-2216使用von Hippel-Lindau (VHL) E3连接酶,且需静脉注射。CRBN结合PROTAC的分子量在711-958 Da之间,亲脂性(pH 7.4下的logD)在1.4-4.8范围内,色谱极性表面积(ePSA)范围为107-146 Ų。图2A显示,口服PROTAC(蓝色区域)大多超出Lipinski的“5法则”(Ro5)和Veber的指南(绿色区域),但氢键供体(HBD)除外。在亲脂性方面,部分PROTAC超过小分子的logP(≤5)和logD(1-3)限制。ePSA的范围较窄,显示其在口服PROTAC设计中更具指导价值。尽管超出传统物理化学限制,口服仍为首选给药途径,除DT-2216和KT-413外,这两者专为血液系统癌症开发。
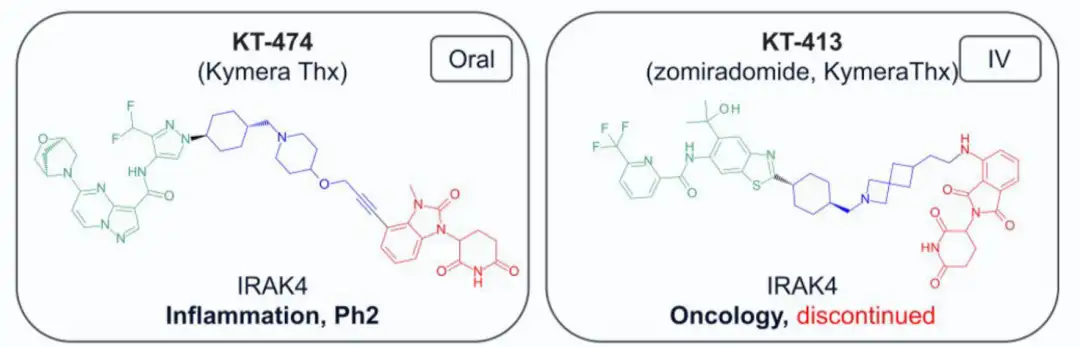
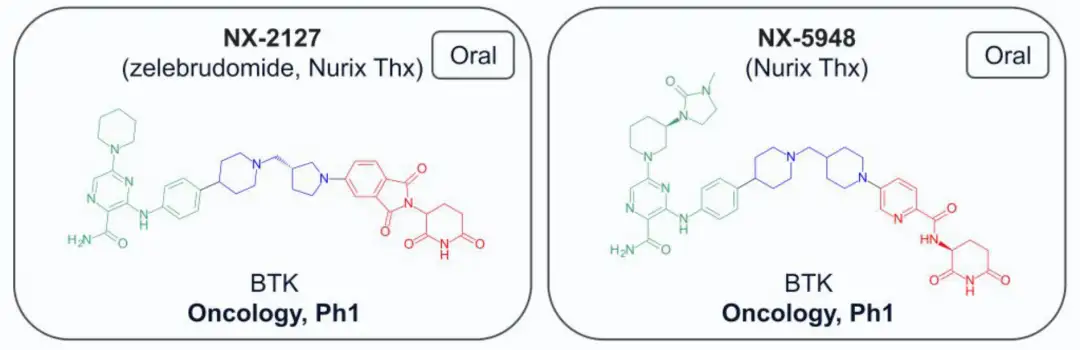
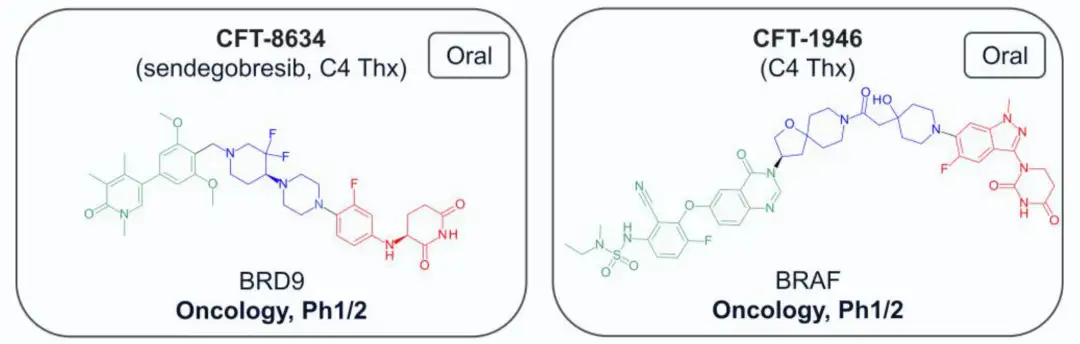
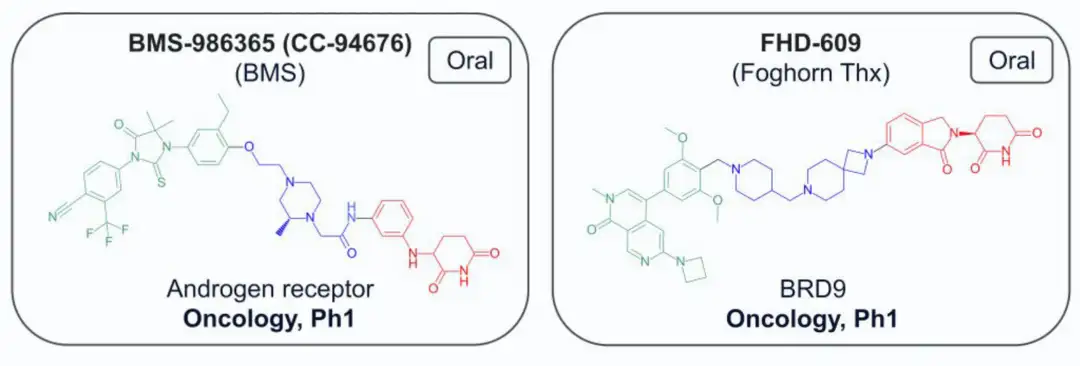
02
Structure of PROTACs
每种临床蛋白水解靶向嵌合体(PROTAC)都经过广泛优化,以在药理和物理化学特性之间实现最佳平衡。PROTAC 的设计包括三个模块化组分——目标蛋白(POI)、E3连接酶配体和连接基,设计技巧在于平衡各个部件的特性,使得最终的 PROTAC 化合物特性整体可接受。
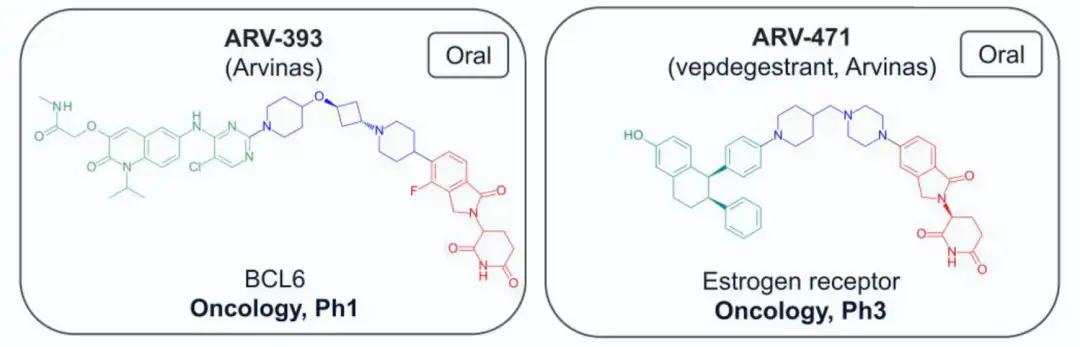
目标蛋白(POI): PROTAC 通常使用对目标蛋白具高效结合力的抑制剂或拮抗剂,多为中性分子并且氢键供体(HBD)数量较少,这样可以避免暴露的 HBD,以利于提高细胞膜透过性。大多数临床PROTAC 的 POI 结合基序溶解性较好,并能连接到 E3 连接酶配体,某些 PROTAC(如 ARV-393)含有少数几个正式供体,但分子内氢键可能屏蔽了这些供体以减少亲水性。
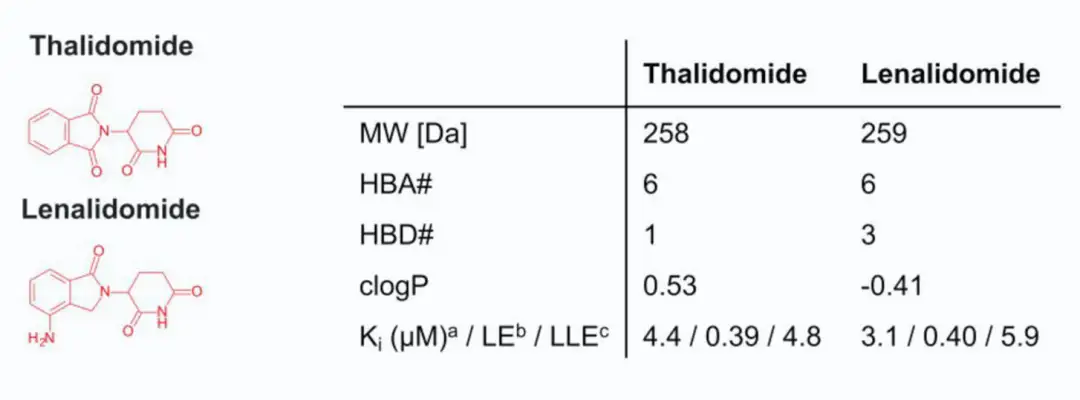
E3连接酶配体: 在临床 PROTAC 中,cereblon(CRBN)是最常用的 E3连接酶,因其低分子量、高极性和高效性成为理想选择。CRBN 配体通常来源于免疫调节药物(IMiD)如沙利度胺(Thalidomide)和来那度胺(Lenalidomide),其主要化学结构包括戊二酰亚胺或二氢尿嘧啶环,具有一个氢键供体且对外排转运蛋白无敏感性。为了应对 IMiD 类配体的安全问题(如致畸性和血液毒性),一些新的 CRBN 配体被应用于 PROTAC,如包含差向异构体混合物的 KT-474 和 CFT-1946 等,其中二氢尿嘧啶基团在 CFT-1946 中展现出改良的化学和代谢稳定性。
连接子: PROTAC 的连接基通常为单个叔胺碱基(例如哌啶、哌嗪或氮杂环丁烷),这些碱性基团通常经过适度调节,以确保 pKa 在合理范围内。对于口服 PROTAC,碱性通常低于预测 pKa 9.1。连接基中多含有环状结构以增加刚性和构象稳定性,大部分临床口服 PROTAC 中至少包含两个环,而在灵活性方面则使用少量亚甲基或乙氧基作为间隔基。例如,CFT-1946 的连接基较为刚性,因其酰胺将哌啶与螺环连接起来,而 KT-474 的连接基较灵活,使用了炔丙基醚间隔基。高刚性的连接基有助于提升靶标降解效力和代谢稳定性,而灵活的连接基则在静脉注射的 DT-2216 中表现较好,DT-2216 的可旋转键数量(29个)远超可接受的范围,但其高度灵活性适合非口服给药。
03
Optimization and Challenges
口服 PROTAC 的生物利用度设计面临显著挑战。由于 PROTAC 的三部分结构,使分子量低于约 700 Da 的设计难以实现,这通常违反了 Lipinski 的分子量规则,同时保持其他 Ro5 标准(如 HBA、HBD、logP)和物理化学描述符(如 PSA、可旋转键)在可接受范围内也具有难度。尽管如此,PROTAC 已在临床前研究和人体实验中显示出一定的生物利用度,能够达到有效的药物暴露水平。
随着 PROTAC 的不断探索,药物化学家积累了关键的物理化学参数数据,提供了新的设计见解。Arvinas 和 AstraZeneca 的研究表明,口服吸收的关键在于减少溶剂暴露的氢键供体(eHBD)数量,两者均独立得出 eHBD≤2 的设计标准,即便分子量、亲脂性、极性和 HBA 等物理化学性质超过了传统的 Ro5 限值。相关研究表明,对于大环 bRo5 药物,酰胺类 HBD 的数量应不超过 2,但正式 HBD 数量可更高,总计不超过 7 个。CRBN 结合基序的小分子特点也符合 eHBD≤2 的标准,这种设计使口服 PROTAC 达到有效生物利用度成为可能,尽管面临挑战。合理分配“分子特性预算”,尤其是控制 HBD 数量,是 PROTAC 设计成功的关键因素。
The three-body problem
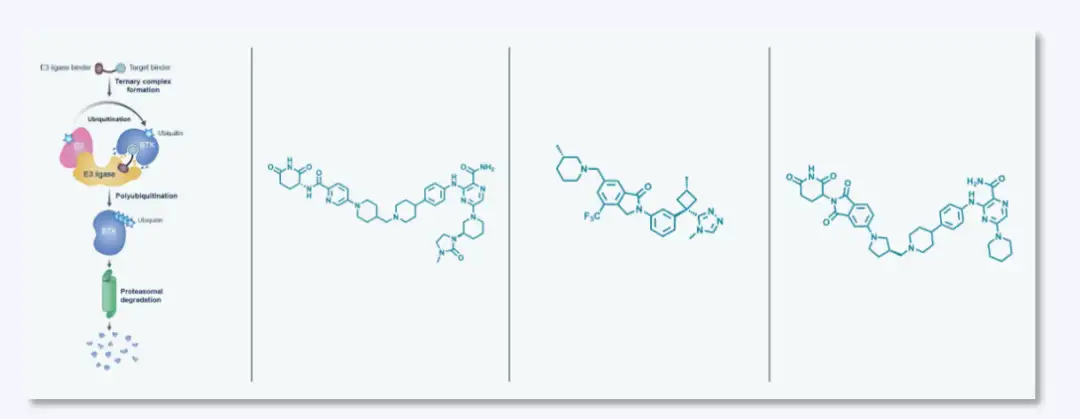
PROTAC 的靶向蛋白质降解功能依赖于三元复合物的有效形成,这种复合物能够促进表面赖氨酸的泛素化。然而,由于可能存在多种构象且并非所有构象都能导致降解,三元复合物的建模十分具有挑战性。PROTAC 的大型分子结构及 POI-linker-E3 组分的相互作用,使得其设计需要更高的“化学公平性”,以确保活性。鉴于形成有效三元复合物的标准仍不明确,生成活性 PROTAC 需要克服设计与合成的重重障碍。
为应对这一挑战,药物化学家已采用“库”的生成方法,通过广泛的化学空间探索,以经验性手段找到可行的组合。开发“PROTAC 工具箱”也提供了一种灵活的筛选方法,包含多种 E3 连接酶、配体和接头的组合,以找到理想的异双功能分子。活性 PROTAC 随后需进一步优化以提升降解效率及改善物理化学性质。当前的化学研究表明,PROTAC 中的线性接头逐渐转变为环状、构象受限版本,以平衡溶解度和细胞渗透性,这也成为设计的共同趋势。
为提高筛选效率并减少浪费,研究人员还开发了“直接生物学”或“纳米 SAR”方法,以便在细胞中直接筛选纳摩尔级的非纯化库,为 PROTAC 的快速、高效和经济可持续的发现提供了新思路。
Shape shifting chimeras
PROTAC 的三部分组成和分子大小使其在构象理解上比传统小分子更具挑战性。药物化学中常用的策略是通过预先组织分子以采取生物活性构象,从而减少结合时的熵惩罚。然而,PROTAC 的配体由于具备较大的构象自由度,加之三元复合物周围的构象可塑性和不确定性,使得模仿哪种“生物活性”构象成为一大难题。
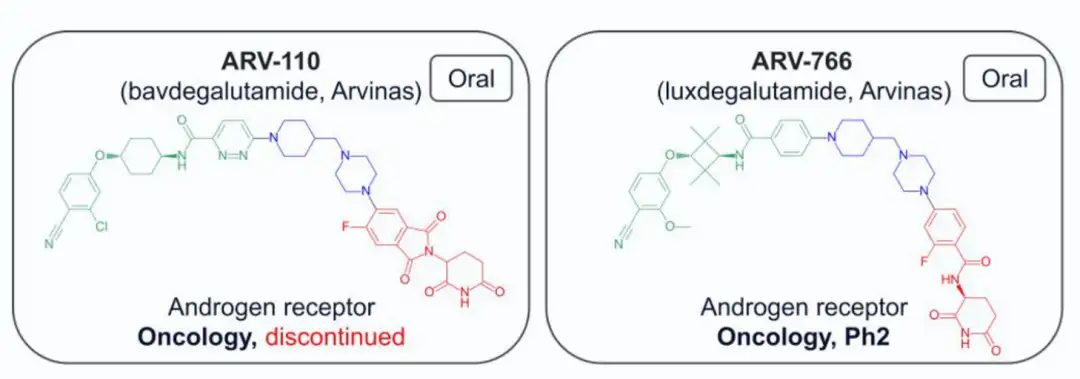
结合 NMR 技术和分子动力学(MD)模拟的研究表明,PROTAC 的构象行为和渗透性在极性(如重水或 DMSO-d6)与非极性溶剂(如 CDCl3)中存在差异。这些差异揭示了构象集合、端到端回折及潜在分子内氢键 (IMHB) 的信息,被称为“变色龙”分子内折叠。与未折叠的分子相比,折叠状态的 CRBN 和 VHL 结合 PROTAC 显示出更高的被动细胞渗透性。而对具有更刚性连接基的 PROTAC(如 ARV-110、ARV-766、ARV-471 和 KT-474)的研究则表明,在不同溶剂中,延伸的构象异构体更为稳定。
The world must be measured by eye
PROTAC 领域的一个关键挑战在于,传统小分子优化程序的体外检测方法是否能为 PROTAC 提供有意义的数据。这些较大的、亲脂性较强的分子易附着在实验器皿、蛋白质或膜上,导致渗透性和蛋白结合检测中的回收率不佳。同时,传统的摇瓶法难以检测出 logD 值大于 4 的化合物,而溶解度也往往低于检测限,使得缺乏数据的候选物分类变得更具挑战性。尽管如此,代谢稳定性测试(如肝细胞和微粒体孵育)对筛除不稳定的 PROTAC 提供了有价值的指标。
为改进 PROTAC 的优化,研究者已借鉴 bRo5 领域的经验,通过色谱法(ChromlogD 和 ePSA)拓展了测量的亲脂性和极性范围,并结合计算机模拟来生成相关的 3D 描述符,从而提升设计成功率。此外,在 FaSSIF 和 FeSSIF 等生物相关溶液中的溶解度测量和“预饱和”法为游离水平的估计提供了更准确的方式。尽管 PROTAC 的亲脂性较传统小分子药物(logD 1-3)更高(13 种临床 PROTAC 中 9 种的 logD≥3),但最终的药代动力学特征仍需通过体内评估在高等动物模型、毒性研究乃至人体中进一步验证。
Pitfalls to avoid
将 CRBN 结合实体(尤其是 IMiD)整合到 PROTAC 中带来了潜在的安全风险。这些风险源于 PROTAC 可能充当分子胶水降解剂 (MGD),引发多种蛋白质(新底物)降解,导致毒理学效应。例如,新底物 GSPT1 是一个翻译终止因子,其降解可能会降低蛋白质合成率并引发细胞毒性,而误导性地掩盖目标 POI 的实际降解情况。此外,不仅 CRBN 配体具有 MGD 活性,其他 E3 连接酶如 VHL 的配体也表现出类似特性。一些 PROTAC 甚至显示出源于 POI 配体的 MG 活性,如一款 JQ1 衍生的 PROTAC 就通过 DCAF16 而非预期的 DCAF15 降解 BRD4,凸显了严格控制实验的重要性,以确保 PROTAC 的机制安全有效。
在发现阶段到体内评估过程中,理解 PROTAC 的代谢命运同样至关重要。即便代谢稳定的 PROTAC,其代谢产物往往更具极性和较低亲脂性,可能降低三元复合物的有效性,进而削弱体内疗效。此外,尽管精心设计避免 MGD 活性,CRBN 结合 PROTAC 的代谢物仍可能具备 MGD 活性,从而带来潜在安全风险。因此,当前临床试验对这些风险的评估将帮助确定 CRBN 基 PROTAC 是否适用于非肿瘤适应症。
04
Conclusions
PROTAC 模式已实现了其最初的靶向蛋白质降解的承诺,多个高质量的降解剂已进入临床评估阶段,尤其是在肿瘤治疗领域,并以 CRBN 为首选 E3 连接酶。初步数据表明,PROTAC 在人体中具备生物学效能和口服生物利用度,进一步验证了其应用潜力。在结构已公开的 PROTAC 中,常见的结构特征如碱基和富环连接子,以及 eHBD≤2 等物理化学参数,为设计提供了宝贵的参考。
PROTAC 以及其他大分子候选药物的出现,挑战了传统 Ro5 规则,表明某些规则可以被重新审视甚至突破。与类似的创新性例子一同,PROTAC 正在重新定义“口服生物可利用化学空间”,表明在探索新领域的边界时,应保持开放态度,同时避免简单地对化学设计设定新的限制。
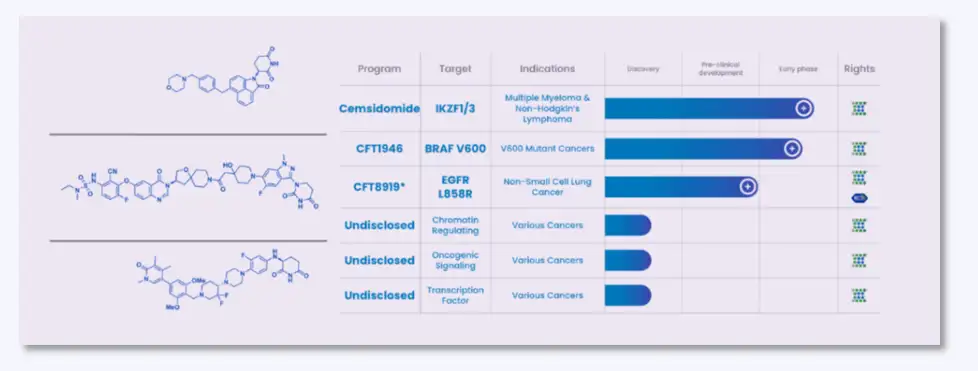
2)C4 Therapeutics处于临床研究的药物及合成
3)Kymera Therapeutics处于临床研究的药物及合成
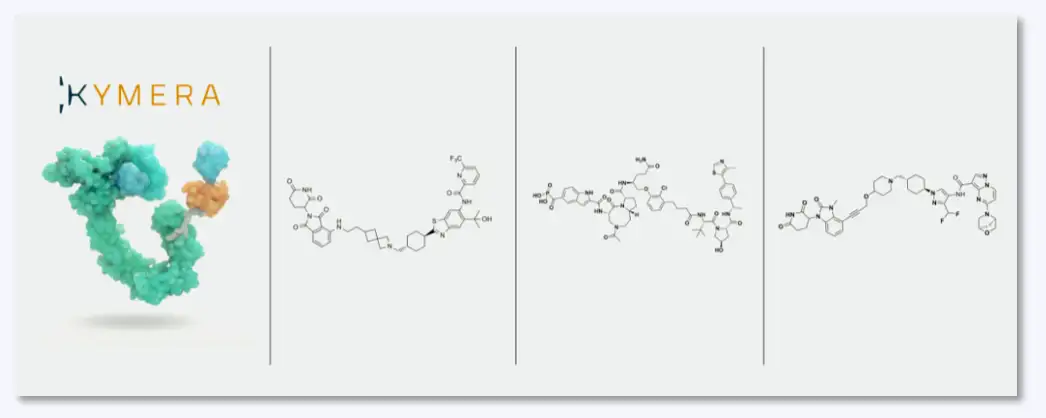
4)Arvinas处于临床研究的药物及合成

 粤公网安备 44010602000473号
粤ICP备09063491号
营业执照
粤公网安备 44010602000473号
粤ICP备09063491号
营业执照

